乐孕途
ART专业学习平台
Cockayne综合征(Cockayne syndrome,CS)是一种世界罕见病,属于常染色体隐性遗传,1936年由英国儿科医生Cockayne博士首次提出,其主要特征是生长障碍、智力缺陷、进行性加重的神经和认知功能障碍以及视力和听力受损,受累儿童可有恶病质及早衰表现中。据统计,美国CS发病率约2.5/1000000,西欧约1/2700000,日本约为1/2770000,国内目前尚没有发病率统计的报道CS主要是由于DNA损伤后转录修复缺陷导致,可使全身多个系统受累,是一种进行性加重的多系统遗传性疾病,通常在婴儿期或者幼儿期开始发病;在新生儿期,大多数CS患儿发育不会受到影响,但因为渐进式的发育衰竭最终会落后于同龄人。目前认为该疾病是由于ERCC8(CSA)、ERCC6(CSB)、ERCC5(XPG)、ERCC3(XPB)、ERCC2(XPD)这5个基因突变所导致。ERCC6(CSB)和ERCC8(CSA)为最主要的致病基因,其中ERCC6基因的突变最常见,约占病例的75%。该基因位于第10(10g11.3)号染色体,可导致CSB型,而ERCC8基因位于第5(5q12.1)号染色体,突变可导致CSA型,约占病例的25%。迄今为止,关于该病的文献报道仍只以个案报道为主。本文报道了1对罕见的同时具有ERCC8基因杂合缺失突变的CSA型携带者夫妇在生育了1个CSA型患儿后,借助胚胎植人前单基因遗传学检测进行阻断,之后成功妊娠的诊疗经过。
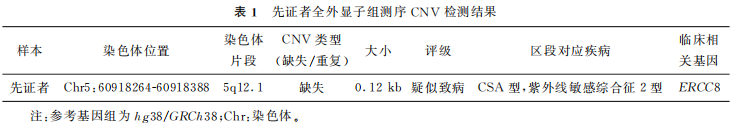
患者夫妇,男方30岁,女方27岁,因生育过1个CSA型患儿,想要借助辅助生殖技术生育1个健康的孩子,于2021年12月27日来甘肃省妇幼保健院生殖医学中心进行孕前咨询。男女双方均为初婚,女方既往孕2产1,2019年药物流产1次,具体情况不详;2018年足月顺产1男孩(先证者),孩子自幼发育落后,8个月会坐,1岁会爬,1岁半会走:协调性差,只会说单字,理解尚可,面部有红色皮疹,对光敏感,四肢肌张力低,小下颌,眼窝稍凹陷,双侧招风耳等。因怀疑某种遗传性疾病,患者夫妇于2021年8月携男孩在北京大学第一医院行全外显子组测序基因拷贝数变异检测(whole exome sequencing for copy-number variants detection,WES-CNV),结果提示患儿5号染色体5q12.1处纯合缺失0.12kb区域,覆盖了ERCC8基因的4号外显子,经家系验证该变异源于夫妇双方,与CSA型和紫外线敏感综合征2型相关,具体结果见表1、图1。
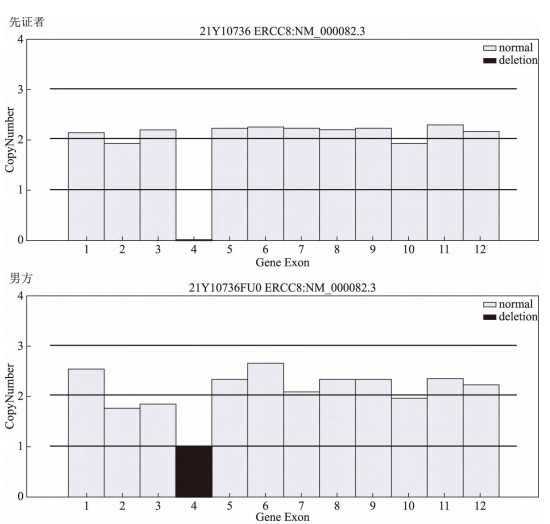
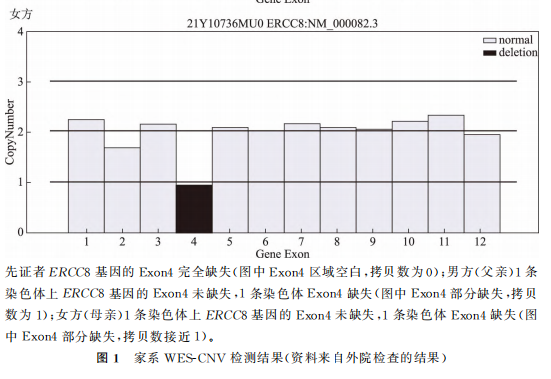
夫妻双方在接受了相应的遗传咨询以及充分获悉相关情况后签署了书面的知情同意书,决定选择PGT-M方案行助孕治疗。此方案经我院伦理委员会审批通过[批件号:(2021)GSFY伦审[65]号]。采集夫妇双方及先证者外周血5ml,对先证者ERCC8基因的Exon4del突变位点进行家系验证。图2为先证者ERCC8基因家系图谱。
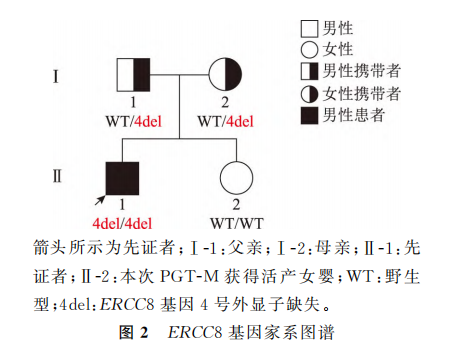
参照美国医学遗传学和基因组学学会(ACMG)指南的评级标准,该突变被判定为可能致病,并由配偶双方分别遗传。同时,对该家系的基因组DNA(gDNA)样本进行目标基因上游和下游的单核苷酸多态性(SNP)多重扩增,以建立家系SNP单体型,为后续胚胎进行SNP连锁分析奠定基础。
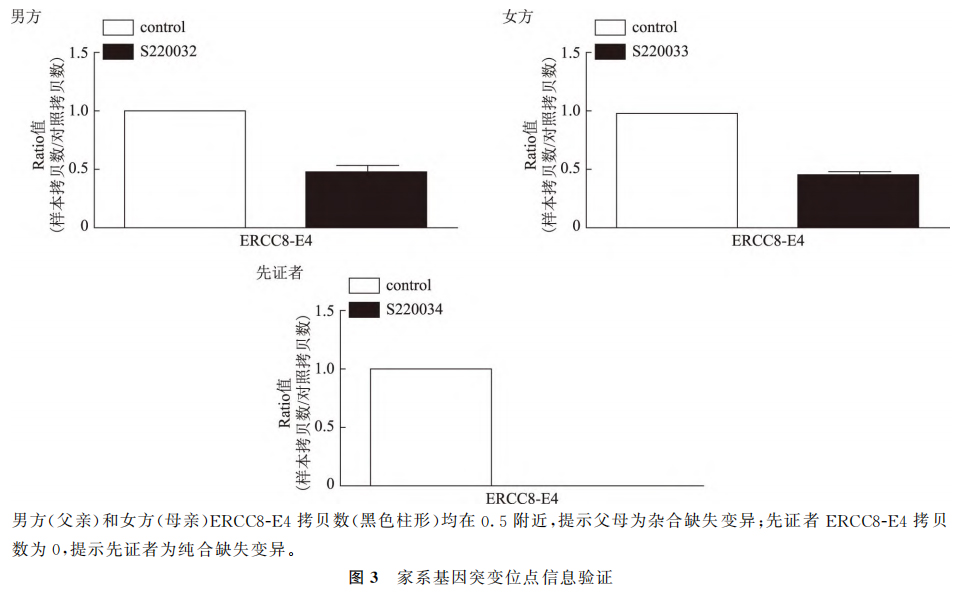
女方采用黄体期短效长方案促排卵,成功获取卵子28枚,采用卵胞浆内单精子注射(ICSI)进行人工授精后有23枚卵子正常受精,形成囊胚21枚,活检囊胚12枚,对活检样本采用单细胞全基因组扩增(Whole genome amplification,WGA),分析胚胎染色体非整倍性。以WGA产物为模板,对ERCC8基因上下游的SNP引物行多重扩增。家系预实验结果如图3所示:ERCC8基因(OMIM:609412)存在小片段的缺失,父母双方均为ERCC8基因杂合缺失变异,先证者分别遗传自父母的基因突变位点:为纯合缺失变异。
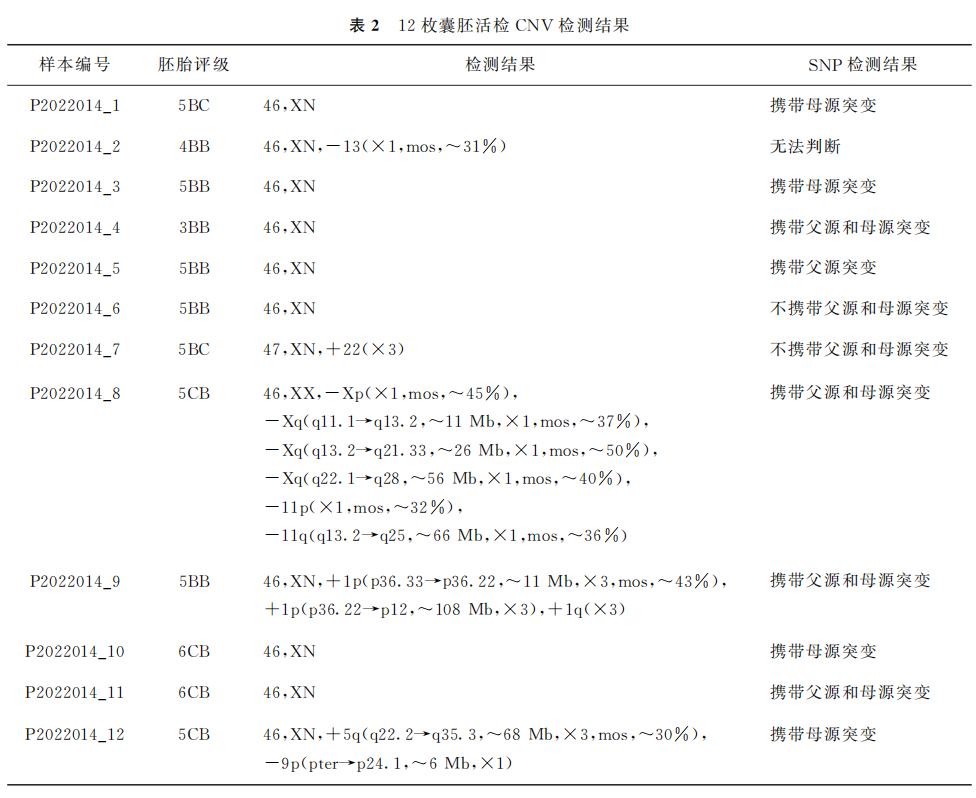
送检囊胚滋养层细胞检测结果示,1枚活检囊胚未检出相关数据,1枚携带父源突变,4枚携带母源突变,4枚为同时携带父源和母源突变的胎,2枚既不携带父源突变也不携带母源突变(表2)。经充分遗传咨询后对患者行冻融胚胎移植,经内膜准备后将编号P2022014_6囊胚(胚胎评级为5BB)移植到母体,最终成功妊娠。妊娠期通过胎儿颈后透明带(NT)扫描、B超检查和羊水穿刺进行胎儿染色体病及单基因病检查证实与前期PGT-M胚胎的检测结果完全相同。女方于2023年1月18日成功分娩1名不携带父源和母源突变的健康女婴,出生时体重3250g。
CS又称为科凯恩综合征、侏儒-视网膜萎缩-耳聋综合征或小头、纹状体小脑钙化和白质营养不良综合征!。患者常常有生长迟缓、发育落后,特殊表现有小头畸形、眼球凹陷、皮肤异常光敏性、可有进行性感音神经性耳聋、进行性视网膜色素变性,还可累积肝脏、肾脏、心血管系统以及内分泌功能。患者大多于20岁之前死亡,且多逝于恶病质引发的呼吸衰竭或者肾脏衰竭。
ERCC8基因位于5号染色体的5q12.1区域,由12个外显子组成,可编码由396个氨基酸组成的DNA切除修复蛋白,可以作为DNA修复因子参与核苷酸剪接修复过程。该蛋白具有色氨酸-天冬氨酸二肽(WD)-40重复序列,包含7个WD结构域,可形成环状β螺旋结构,可作为蛋白质相互作用的支架,并且是转录偶联DNA修复所需的E3泛素连接酶复合物的一部分,在紫外线诱导的DNA损伤的核苷酸切除修复转录偶联亚途径中起重要作用。当ERCC8基因的4号外显子缺失时会影响ERCC8蛋白的WD-40重复元件,从而导致DNA修复缺陷。缺陷时细胞无法修复基因活跃转录链损伤,经紫外线照射后细胞RNA合成恢复率下降,患者表现出皮肤光敏性,短时间暴露于阳光,即可发生不同程度晒伤,出现面部红疹表现。患者还可表现出严重的生长发育障碍和神经发育缺陷。目前认为神经发育表型是转录失调的结果,可能源于DNA损伤的内源性来源“7,但对于转录和核苷酸切除修复障碍仍不能完全解释有关神经系统退化方面的症状。有研究报道,CS的致病机制有:转录和转录偶联核苷酸切除修复缺陷、碱基切除DNA修复缺陷和线粒体功能障碍、异染色质丢失、p53泛素化异常、细胞分裂及端粒维持异常等。根据疾病起病年龄与严重程度,可分为工型(经典型)、Ⅱ型(早发型)、Ⅲ型(晚发型)、脑-眼-面-骨(COFS)综合征、紫外线敏感综合征(UVSS)。极少数患者同时具有CS和着色性干皮病表型,称为着色性干皮病-CS型2]。症状从重到轻依次为COFS综合征、Ⅱ型、I型、Ⅲ型和UVSS。
目前临床上对于CS尚无有效的治疗方法,以对症治疗为主,旨在提高患者的生活质量。有研究表明,现在关于CS治疗的方向主要有:改善线粒体功能、修复DNA损伤、干细胞治疗等,但均处于细胞或动物模型研究阶段,所以目前的干预仍然以预防为主。CSA型为常染色体隐性遗传,且所有CS亚型都有此遗传特点,当配偶双方都携带致病的杂合突变基因时,每次怀孕后代均有25%的可能性为CS患者、50%为无症状携带者、25%为正常人。因此,提倡CS致病基因携带者及CS患者的同胞在生育前积极进行遗传咨询,通过基因检测判断是否为CS致病基因携带者,若为携带者,在生育时,需进行夫妻双方的CS致病基因检测,若夫妻双方均为携带者就可选择PGT-M辅助以降低CS患者的出生。对于基因诊断明确的家庭,若已经自然受孕,为明确产前诊断,在妊娠时母亲一定要对羊水细胞或者是胎盘绒毛细胞进行基因分析。针对ERCC8基因突变导致的CSA型,其突变类型多数为点突变;ERCC8基因的Exon4缺失在CS患者中少有报道,且夫妻双方均为Exon4缺失的尚未见报道。
单基因遗传病,多数可致畸致死且具有家族性和终生性的特点,患者常有严重的临床表现,社会参与性差,对患者家庭及社会造成严重负担。虽然单基因病的发病率低,从几千分之一到几千万分之一.但是种类繁多,是出生缺陷的主要原因之一。PGT-M技术,避免了存在单基因病风险的胚胎植入母体,能更好地实现优生优育,现已广泛应用于单基因病的孕前阻断,已经成为预防单基因遗传病的重要手段之一。本研究中的患者夫妇通过PGT-M技术,对植入母体内的囊胚进行提前遗传性疾病相关检测,选择同时不携带父源突变与母源突变的胚胎进行移植,最终获得活产,即CSA型在该家系已被成功阳断。对罕见病的罕见基因突变进行生育指导在临床工作中具有非常重要的意义,本报道旨在通过回顾该对患者夫妇的诊疗经过为临床中单基因病患者的治疗决策提供一些思路,为PGT-M技术在临床工作中的应用提供更多数据积累。